The CompBioMed effort against coronavirus aims to accelerate the development of antiviral drugs by modelling proteins that play critical roles in the virus life cycle in order to identify promising drug targets. Our proposed work will develop machine learning (ML), deep learning (DL) and artificial intelligence (AI) techniques to:
1) identify and build accurate three-dimensional structural models of the SARS-CoV-2 proteome by closely integrating experimental structural and systems biology datasets,
2) accelerate adaptive conformational sampling of the viral proteins to potentially identify novel binding sites/pockets that can be targeted by small molecules,
3) rapidly filter, rank, and search for small molecules across widely available chemical libraries, and to integrate virtual screening (computational drug discovery techniques) techniques with experimental high throughput screening, and
4) enable multi-scale, multi-resolution simulations of the SARS-CoV-2 viral envelope, and specific proteins.
Across various supercomputing facilities in the US and Europe, three collections of drug candidates are being screened for inhibitor activity:
1) Known and licensed drugs for quick repurposing opportunities (e.g., DrugBank),
2) Library of 100M known small molecules that are drug like (e.g., PubChem) and
3) Large-scale libraries (e.g., Enamine, ZINC) with billions of compounds that could be manufactured quickly for testing.
Our primary targets are existing drugs that are currently in manufacturing pipelines and can be repurposed quickly. The first of our preliminary results are coming in and will be made available to community wet labs in the coming days for experimental testing and screening.
Our multi-level screening approach first uses fast machine learning and docking methods to provide an initial ranking of drug compounds. This ranking is refined by using methods that take the protein response into account as well as the initial protein states. These methods include machine learning driven sampling as well as accelerated dynamics methods to sample the protein and protein-ligand complex phase space. For the accelerated dynamics methods, we are deploying capabilities developed in the latest versions of Amber as well as MD capabilities in BER’s NWChem package. The CCS group will carry out more advanced binding free energy prediction calculations on thousands of compounds selected by this workflow. These computationally intensive calculations will be carried out the supercomputers Summit, SuperMUC-NG, and Scafell Pike. Use of these supercomputers has been enabled by the unique collaboration between the consortium on coronavirus and CompBioMed2.
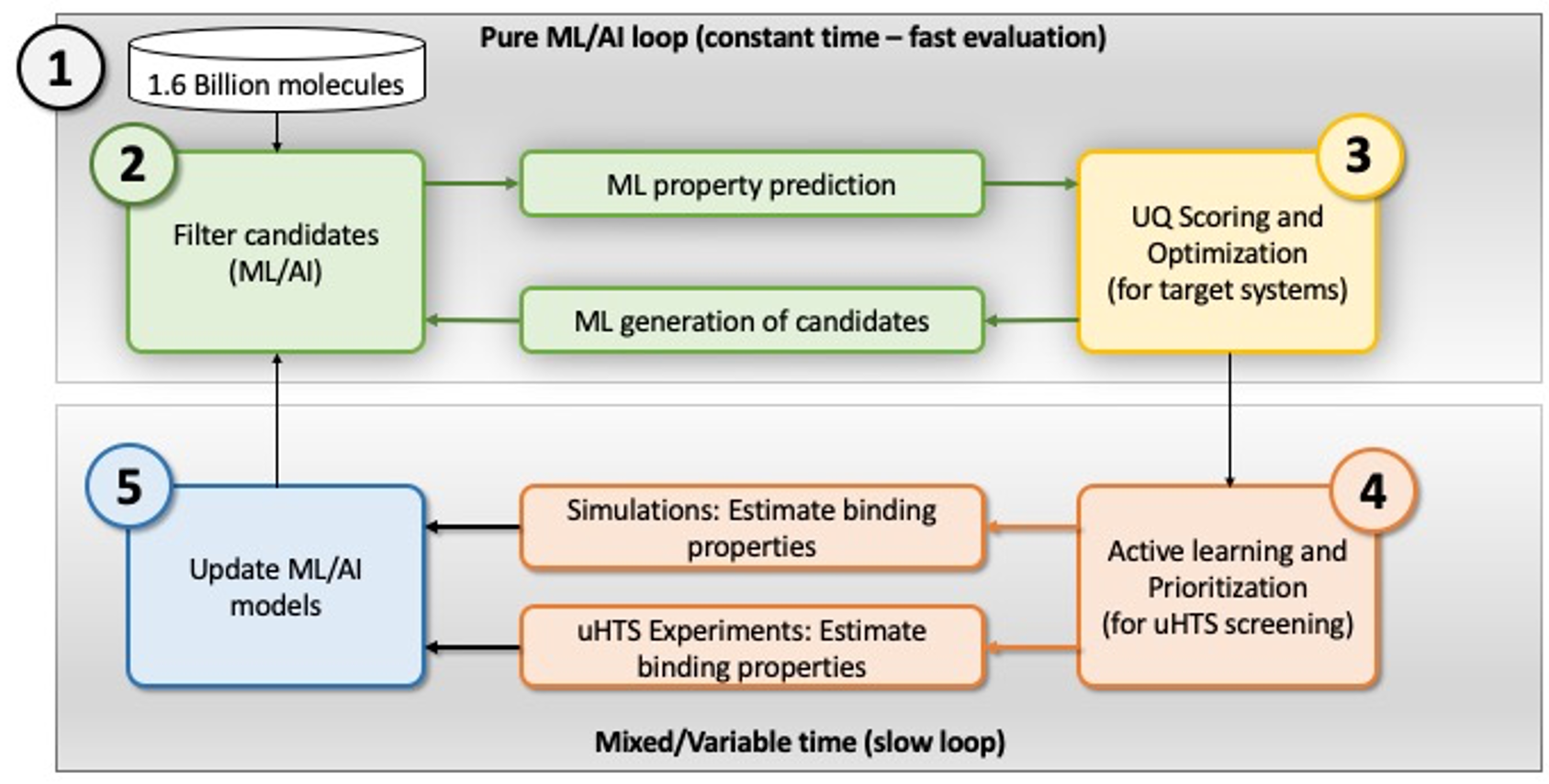
A schematic of the closed loop active learning strategy identify compounds that can be used as leads for COVID-19 countermeasures.
Prof Coveney’s CCS at UCL is seeking to identify promising inhibitors of COVID-19 targets through assessing the potential to reposition existing drugs in combination with machine learning alongside so-called deep-drive applications of molecular dynamics and artificial intelligence methods. We are interested in additional contributors to this effort, in particular from those with expertise in machine learning and generative methods for compound discovery, and physics based methods for calculating binding free energies. If you wish to contribute, please get in touch by emailing Dr Hugh Martin (h.s.martin “at” ucl.ac.uk) with your name, institution, a one-paragraph summary of your area of expertise, and up to 5 of your most relevant publications, making clear which of the 6 areas of coronavirus research you are seeking to contribute to in this consortium.
CompBioMed-Coronavirus Pages:
CompBioMed and Coronavirus
The Consortium on Coronavirus
CompBioMed Partner Activity
Call for Contributions
Coronavirus Research Resources
Coronavirus Blog
Areas of Research:
Computational Drug Discovery
Epitope Analysis
Drug Toxicity
Computational Epidemiology
Virus Evolutionary Analysis
Host Response Analysis