For clinical visitors to our website, we provide on this page selected CompBioMed links and services of particular interest to you. Please check out the links on the column to the right, and the case studies featured below.
We interviewed a number of clinicians working with our researchers in September 2019 to ask them about their experiences of CompBioMed and how it has helped their work. This can be viewed on our YouTube channel and below:
If you are interested in how our simulations can help in your work, we have also provided succinct descriptions of our models. Click on the boxes for more information or go to our Software Hub
 |
 |
|
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
||
 |
 |
 |
CT2S: a template of high-throughput in silico medicine services for clinical research and specialist care
Osteoporosis (progressive loss of bone mass) and increased risk of falling are the primary cause of the so-called fragility fractures, bone fractures that occur with minor traumas that in a normal healthy subject should not cause any fracture. According to the International Osteoporosis Foundation approximately 1.6 million fragility hip fractures occur worldwide each year, but by 2050 this number could reach between 4.5 million and 6.3 million. Subjects considered at general risk are referred to a specialist, who decide whether to prescribe a life-long pharmacological treatment (antiresorptive drugs), or to check the patient again in a few years. This decision is currently based primarily on an instrumental value, called T-score, which indicates how lower is the bone mass of the patient compared to that of a healthy population; patients are considered osteoporotic if their T-score is less than -2.5. However, about half of the patients who experience a fragility hip fracture, have a T-score greater than -2.5, which may appear a paradox. In reality the risk of hip fracture is linked to the strength of the patient’s femur, but this is something that cannot be measured non-invasively.
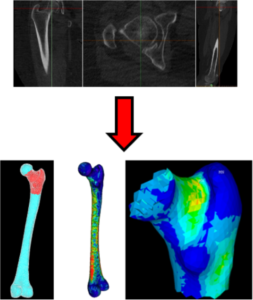 Prof Marco Viceconti has worked on the prediction of femoral strength from medical imaging data for over ten years, first at the Rizzoli institute in Italy, and more recently at the Insigneo institute in Sheffield. The most recent computational medicine technology, code-named CT2S can predict the strength of a human femur from properly calibrated Computed Tomography images with an accuracy of 84-85% [Viceconti et al., 2018]. When used to separate the patients at high risk of hip fractures from those a lower risk, CT2S accuracy is very close to this level of accuracy (82-83%) [Viceconti et al., 2018]. In a recently published study [Viceconti et al., 2018], it is concluded that the CT2S technology is immediately cost-effective for clinical trials using bone strength as end point, while it would become cost-effective if the price-point per simulation would be reduced. Interestingly, a similar study on a similar technology aimed to predict the risk of vertebral fractures, reached nearly identical conclusions [Agten et al., 2017].
Prof Marco Viceconti has worked on the prediction of femoral strength from medical imaging data for over ten years, first at the Rizzoli institute in Italy, and more recently at the Insigneo institute in Sheffield. The most recent computational medicine technology, code-named CT2S can predict the strength of a human femur from properly calibrated Computed Tomography images with an accuracy of 84-85% [Viceconti et al., 2018]. When used to separate the patients at high risk of hip fractures from those a lower risk, CT2S accuracy is very close to this level of accuracy (82-83%) [Viceconti et al., 2018]. In a recently published study [Viceconti et al., 2018], it is concluded that the CT2S technology is immediately cost-effective for clinical trials using bone strength as end point, while it would become cost-effective if the price-point per simulation would be reduced. Interestingly, a similar study on a similar technology aimed to predict the risk of vertebral fractures, reached nearly identical conclusions [Agten et al., 2017].
In the CompBioMed project the CT2S technology has been deployed on the ShARC HPC cluster at the University of Sheffield, and optimised so that the simulation now runs over 20 times faster than on a standard workstation. A web service portal has been deployed to allow clinical researchers across the world to access this powerful computational medicine tool as part of their clinical trials.
Now we are working on a new version of the CT2S service, called CT2S-HT designed to provide the level of automation required in high-throughput clinical studies, in routine clinical use, and in new in silico-augmented clinical trials where thousands of virtual patient models need to be solved. Large volumes of CT datasets, their segmentation, and their QCT calibration, will be stored in the Insigneo secure XNAT server; from there the CT2S-HT service will automatically generate the patient-specific stochastic finite element models, solved them on the ShARC HPC system, extract the relevant results, and stored them back in the XNAT server, properly linked to the patient ID. By the end of the CompBioMed project, in 2019, we expect to have the CT2S technology exposed as a low-cost high-throughput service, cost-effective for widespread clinical use.
This work was carried out by Marco Viceconti, Pinaki Bhattacharya, Will Griffiths, Sachin Ramanatha, and Shannon Li at the Department of Mechanical Engineering and Insigneo Institute for in silico Medicine
References
- Agten CA, Ramme AJ, Kang S, Honig S, Chang G. Cost-effectiveness of Virtual Bone Strength Testing in Osteoporosis Screening Programs for Postmenopausal Women in the United States. Radiology. 2017; 285:506-517. https://doi.org/10.1148/radiol.2017161259
- Viceconti M, Qasim M, Bhattacharya P, Li X. Are CT-Based Finite Element Model Predictions of Femoral Bone Strengthening Clinically Useful? Curr Osteoporos Rep. 2018 Apr 14. https://doi.org/10.1007/s11914-018-0438-8
Predicting if individual bacterial protein mutations confer antibiotic resistance.
Everyone appreciates the threat to modern medicine posed by the rise of antibiotic resistance and the resulting urgent need to develop new classes of antibiotics. What is less well understood is that new, faster diagnostic methods are at least as important as new drugs in sustaining our ability to treat bacterial infections.
The traditional approach to determining which antibiotics can be used to treat an infection is to grow (culture) a sample and then expose it to a panel of antibiotics and determine which prevent further growth. Whilst there have been important advancements in imaging and automation, Alexander Fleming would have understood this method.
There is, however, a coming technological revolution in clinical microbiology. One of the most promising approaches is to sequence the whole genome of the infecting pathogen and, having compared to a reference genome, look up any encountered mutations in a panel of key genes and infer their effect on a panel of antibiotics. This is potentially faster, more reproducible, is getting cheaper all the time, and you also get epidemiological data for free.
Nor is this fantasy; since March 2017, all patients with suspected Tuberculosis in England have routinely had their sputum sample sequenced and an antibiogram inferred by Public Heath England [1]. Such a whole-genome sequencing (WGS) clinical microbiology service has one key weakness; it cannot return a result for mutations that have not been encountered enough times to allow them to be classified.
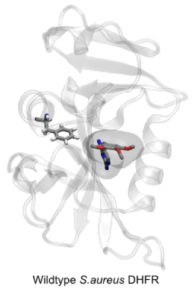 Researchers at the Oxford NIHR Biomedical Research Centre, led by Philip Fowler, are developing methods using molecular simulation that are able to predict the effect of individual mutations on the action of an antibiotic. The hypothesis on which the method is based is that mutations in a bacterial target protein confer resistance by abrogating the binding of the antibiotic whilst, crucially, not materially affecting the binding of the natural substrate. This boils down to calculating the effect of the mutation on the antibiotic and substrate binding free energies. Since the antibiotic often competes with the substrate this is a difficult and subtle problem and so faster, but less necessarily less accurate, methods of calculating free energies are ineffective.
Researchers at the Oxford NIHR Biomedical Research Centre, led by Philip Fowler, are developing methods using molecular simulation that are able to predict the effect of individual mutations on the action of an antibiotic. The hypothesis on which the method is based is that mutations in a bacterial target protein confer resistance by abrogating the binding of the antibiotic whilst, crucially, not materially affecting the binding of the natural substrate. This boils down to calculating the effect of the mutation on the antibiotic and substrate binding free energies. Since the antibiotic often competes with the substrate this is a difficult and subtle problem and so faster, but less necessarily less accurate, methods of calculating free energies are ineffective.
Philip Fowler presented results from a proof-of-principle study on the action of trimethoprim in S. aureus [2] at the recent CompBioMed & BioExcel Free Energy Workshop at UCL. He showed how large numbers of short replica-exchange thermodynamic integration calculations were able to not only predict which mutations conferred resistance (and which did not) but also that the calculations were reproducible, a necessary condition if the results of this method are ultimately to be included in any clinical microbiology service.
References
- Walker TM, Cruz ALG, Peto TE, Smith EG, Esmail H, Crook DW. Tuberculosis is changing. Lancet Infec Dis 2017; 17: 359–61.
- Fowler PW, Cole K, Gordon NC, Kearns AM, Llewelyn MJ, Peto TEA, Crook DW, Walker AS. 2018. Robust Prediction of Resistance to Trimethoprim in Staphylococcus aureus. Cell Chem Biol 25:339–349
More examples of clinical users:
- CompBioMed e-Seminar #31 - CompBioMed’s 31st e-seminar took place on 26 July 2023, titled “Assessing the Credibility of Computational Models: Application of the FDA-Endorsed ASME VV-40“. Computational models in the medical field are more and more stepping out the door of the laboratories they were developed in to find practical applications in the clinical or regulatory practice, for instance.…
- CompBioMed Conference 2023 – Abstract Submission Deadline Extended to 30 June 2023 - Due to popular demand, the abstract submission deadline for CompBioMed Conference 2023 has been extended to 30 June 2023. You can read more and submit abstracts on the conference website.
- CompBioMed Newsletter Issue 12 – Out Now - The latest issue of the CompBioMed Newsletter is now available, addressing the upcoming CompBioMed Conference 2023, the book Virtual You, the CompBioMed film Virtual Pandemics, our scalability service, and biomedical urgent computing in the exascale era. Click here to view it. To find our other newsletter, follow this link.
- CompBioMed at Le Studium Conference on Cardiovascular Modelling - CompBioMed will be at the Le Studium Conference on “Cardiovascular Modelling: Basic Science to Clinical Translation” to be held in Tours (France) on 13th and 14th December 2022. The symposium is being organised, managed and sponsored by the Le Studium Institute for Advanced Studies – Loire Valley and the INSERM Unit iBrain Imagerie et Cerveau.…
- CompBioMed Session at VPH2022 - The CompBiomed centre of excellence organised, in collaboration with the VPH Institute, a special session on the The role of exascale computing in computational biomedicine at the VPH2022 conference in Porto (PL) on Friday 9 September 2022. The session included four speakers from our consortium. Mariano Vasquez (BSC) showed concrete examples of how Alya can…