Application description
 HemeLB, developed by the team of Prof Peter Coveney at University College London (UK), is a 3D macroscopic blood flow simulation tool that has been specifically optimized to efficiently solve the large and sparse geometries characteristic of vascular geometries. It has been used to study flow in aneurysms, retinal networks, and drug delivery among many other cases. Target users will be trying to understand blood flow in complex vascular domains where 3D knowledge of the geometry is critical to solving their problem. HemeLB is open-source and available for free download from Github under the LGPL-3.0 License. Build instructions are provided either from the repository itself (via the ReadMe file or using a provided build script) or via the HemeLB website. External users will need to compile the code themselves on their target machine. HemeLB has been successfully built on a wide variety of HPC systems including ARCHER2, SuperMUC-NG, Summit, and Blue Waters.
HemeLB, developed by the team of Prof Peter Coveney at University College London (UK), is a 3D macroscopic blood flow simulation tool that has been specifically optimized to efficiently solve the large and sparse geometries characteristic of vascular geometries. It has been used to study flow in aneurysms, retinal networks, and drug delivery among many other cases. Target users will be trying to understand blood flow in complex vascular domains where 3D knowledge of the geometry is critical to solving their problem. HemeLB is open-source and available for free download from Github under the LGPL-3.0 License. Build instructions are provided either from the repository itself (via the ReadMe file or using a provided build script) or via the HemeLB website. External users will need to compile the code themselves on their target machine. HemeLB has been successfully built on a wide variety of HPC systems including ARCHER2, SuperMUC-NG, Summit, and Blue Waters.
Technical specifications
HemeLB is written in C++ and uses MPI to distribute work amongst multiple processors. It has been designed to conduct a single run of a flow problem in a monolithic format. Aside from a compiler and MPI library, HemeLB comes packaged with the dependencies necessary for building and running the code. One dependency (CTemplate) currently requires a version of Python2 for successful compilation, however, we are in the process of transferring this to Python3 as it becomes essential on different HPC platforms. Versions of HemeLB that are accelerated by GPUs are currently being developed. Those currently/soon-to-become available utilise CUDA to enable acceleration on NVIDIA GPUs. A port, using HIP to enable execution on AMD GPUs, has been conducted and is currently being tested. As a rule of thumb, input files and simulation data scale linearly with the number of sites being simulated. Execution may require available memory of around 2kB/lattice site.
HemeLB geometries can be developed using the tools available at https://github.com/UCL-CCS/HemePure_tools, whilst post-processing of results generally makes use of HemeXtract to convert the binary output files to a human-readable format. These can then be viewed with scientific visualization software such as ParaView.
CompBioMed partners UCL and LRZ have worked on a video (link) visualising the blood flow in the human forearm and how this was created with the HemeLB application. This work will be shown as part of the "Scientific Visualization & Data Analytics Showcase" at 2021 supercomputing conference, SC21, and has been shortlisted as one of the five best scientific visualisations in the conference programme.
HPC usage and parallel performance
Simulation of blood flow in the full human vasculature is a challenging task for any computational approach. Although 1D solvers can do this with less computational resources than a 3D model it does not provide the same level of fundamental flow details and personalization of the flow domain. For the development of virtual human simulations, high fidelity 3D simulations – such as those generated by HemeLB – will be crucial to both medical and scientific understanding. The efficient generation of such simulations will make the use of HPC essential, this is the only way that flow of physiologically relevant timeframes can be created within a reasonable simulation time. At the full-human scale, where a high-resolution decomposition of the vasculature could require more than 50 billion lattice sites, such simulations can demand the resources of an exascale machine.
HemeLB manifests excellent strong scaling results on a number of HPC platforms. Here we highlight the performance of the CPU version of the code on SuperMUC-NG (LRZ, Germany) and the NVIDIA GPU version on Summit (USA) – machines at #17 and #2, respectively, on the Nov 2021 Top500 list. To compare GPU and CPU results, we convert the number of GPUs to CPU equivalent by using the number of streaming multiprocessors available on the chip – this is the methodology used by the Top500 list.
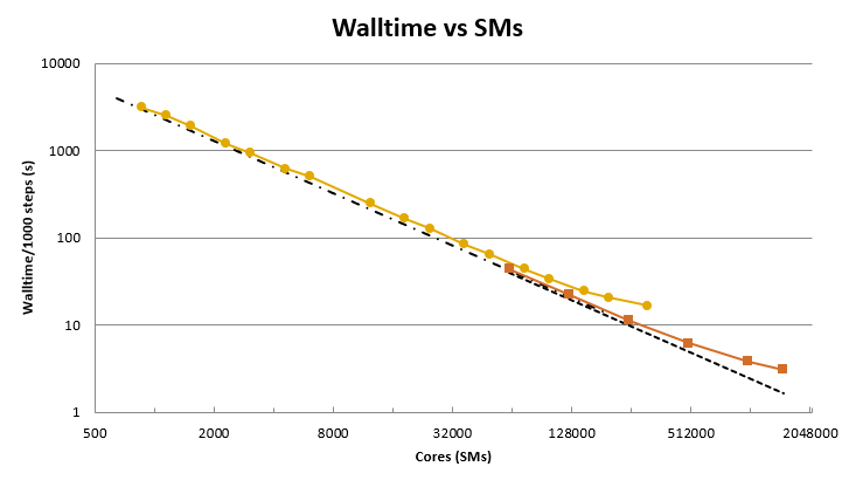
In the Figure above we present our current observed strong scaling behaviour with HemeLB. These have been obtained using a 10 billion site circle of Willis domain that represents the complexity of the domain used in production jobs. The data has been scaled to cores for the CPU code and SMs (i.e., CPU core equivalent) for the GPU data. From these plots, it can be seen that HemeLB demonstrates very good strong scaling behaviour to the full production partition of SuperMUC-NG (approx. 310,000 CPU cores, the fastest CPU only machine in Europe). On Summit, the use of up to 20,000 GPUs allows this to be extended up to approx. 1.5 million SMs. In further testing, we have been able to extend execution with acceptable performance up to 2 million SMs. These last two figures demonstrate that HemeLB is already able to effectively run at around the maximum day-to-day usage levels expected of an exascale machine.
Within a collaboration with FocusCOE, UCL has also developed a dedicated training website for users to get to support with the deployment and benchmarking of HemeLB. The training is designed with the purpose of acquiring performance data and scaling information on emerging exascale architectures.
HemeLB uses MPI in a master-worker format to manage communications across the assigned resources. Here a single rank is dedicated to managing the simulation whilst the remainder are devoted to conducting the computation throughout the domain. Communication between ranks occurs when a lattice site’s neighbours are being computed on a separate rank. MPI communications have been organized to mask this as much as possible. Data input all occurs within the initialization phase of the simulation, this can correspond to a significant quantity of data – approximately 1.3kB/site. Data output occurs at a frequency indicated by the user in the input file, this output will typically require 64 bytes/site (though variable dependent on actual output requested) and this is stored in a compressed binary format. Checkpoint files can also be saved in this manner if required. For the single component version of HemeLB discussed here, no temporary files are written to aid simulation completion.
Within CompBioMed2, we have achieved a number of technical developments for the HemeLB code and are continuing to work towards several more. The port to NVIDIA GPUs is the primary exemplar of this, resulting in the strong scaling results presented above. Efforts within CompBioMed2 have also developed a port for AMD GPUs and the performance of this is currently being evaluated. Within the next 12 months, it is possible that similar work will be undertaken to enable use on Intel GPUs (such as being used in LRZ’s SuperMUC-NG Phase 2). Whilst platform-agnostic programming is an ideal goal, it will take time before a consensus is reached on how this is to be best achieved for GPUs (if possible). Further optimization work (current and ongoing) with LRZ has been focused on improving the performance of HemeLB on SuperMUC-NG. This has included an update of the intrinsics used to accelerate key computational kernels within the code – this effort has demonstrated improvements to the parallel efficiency of the code and we are currently working to evaluate the large-scale performance of these. Ongoing work will further look at how memory management could be optimized for this machine. Whilst all of these could be described as co-design, further explicit co-design efforts are currently in the planning stages for the possible use of HemeLB as benchmark code for testing, evaluating and commissioning new hardware such as NVIDIA’s GPU hardware development kits (at UCL), SuperMUC-NG Phase 2 (at LRZ) and the upcoming European Processor (with ATOS).
As part of the work in CompBioMed, EPCC have developed a service called the HemeLB High Performance Offload service (Hoff) which allows to offloads HemeLB simulations to a remote HPC system, using a set of REST endpoints for submitting and managing computational jobs on remote resources. With the Hoff, existing Portals or existing SaaS offerings are able to upload its input files to a server connected to the HPC system where the simulation will be executed. After copying the input data sets onto the target HPC system, the Hoff schedule and execute the simulations, and, once completed, notify the user and allow for retrieve of the output data. The tool is currently interfaced between a portal, PolNet, which extents the functionality of HemeLB, with the Cirrus HPC system, and EPCC is currently working to extend its functionalities to work with the SLURM scheduling system.
Clinical Use:
Digital Twin
License type:
Open source (LGPL) on GitHub, free
User Resources
Related articles
- Groen D et al. 2013, Analysing and modelling the performance of the HemeLB lattice-Boltzmann simulation environment. DOI
- Nash RW et al. 2014, Choice of boundary condition for lattice-Boltzmann simulation of moderate-Reynolds-number flow in complex domains. DOI
- Mazzeo MD, Coveney PV 2008, HemeLB: A high performance parallel lattice-Boltzmann code for large scale fluid flow in complex geometries. DOI
- Mc Cullough JWS et al. 2021, Towards blood flow in the virtual human: efficient self-coupling of HemeLB. Interface Focus 11: 2019 01 19. DOI