PlayMolecule is a drug discovery web service. It contains a variety of applications which allow users to accelerate and improve their drug discovery workflows using novel machine learning methods (such as binding affinity predictors) or through molecular dynamics simulations to elucidate biological structures and binding modes.
PlayMolecule is used daily by academic institutions and industry. Both students, as well as experienced researchers, are using PlayMolecule to evaluate machine learning methods or simplify their molecular dynamics workflows. PlayMolecule already counts close to two thousand registered users with around 80 new users registering every month.
PlayMolecule is available at https://playmolecule.com/. The free access has restrictions imposed on atom count of molecular systems due to constraints in computing resources which are available.
Technical specifications
PlayMolecule consists of various separate components. The web server, the backend server and the individual applications. The web server is written in Python, React and Angular, the backend is written in GoLang and the applications are written mostly in Python with some lines in C++. The web server and backend server have a total memory requirement of around 12 GB RAM. The individual application runs usually require 16 GB or more RAM, at least 2 CPU cores and a few of them relating to molecular dynamics and neural networks require NVIDIA GPUs. The code is deployed with a custom Python installation script which handles all the system setup and data shipping from Google Cloud Storage.
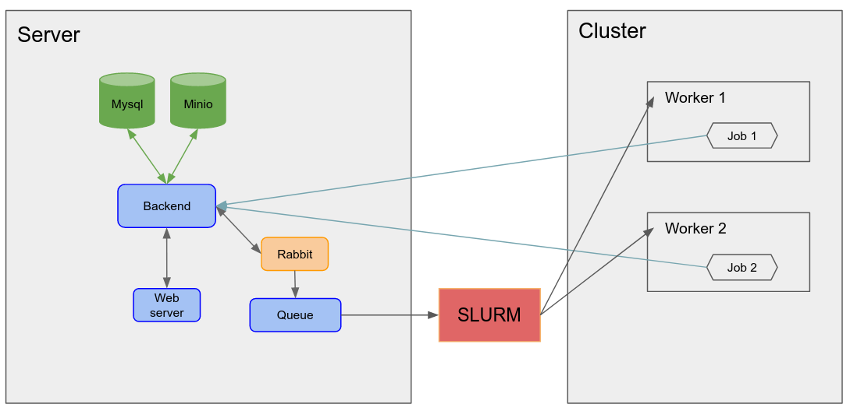
The architecture of PlayMolecule can be seen in the figure above. The web server communicates with the computation backend (written in GoLang) which in turn sends job executions through RabbitMQ to the Queue app which communicates with the queueing system. The queueing system then distributes the jobs to the workers and once completed the jobs send their results back to the backend which stores them in the MinIO server. The backend also employs a MySQL database to manage users, job executions, MinIO file tags and other information necessary.
The code itself in the PlayMolecule backends is mostly serial (excluding the Apache server). However individual jobs are typically sent to a queueing system such as SLURM and can run in an embarrassingly parallel manner. All PlayMolecule applications are containerized with Singularity to ensure easy deployment and reproducibility. For visualisation of the user interfaces and the results, PlayMolecule offers a web server which integrates a state-of-the-art 3D molecular viewer.
HPC usage and parallel performance
HPC resources can be leveraged by PlayMolecule both to serve a larger number of users as well as to provide faster computation. Applications such as AdaptiveSampling depend on multiple GPUs being available so that multiple simulations can be run in a high-throughput parallel manner to explore faster protein conformational space, or protein-ligand interactions and thus can fully leverage large computing clusters.
PlayMolecule applications mostly run in an embarrassingly parallel manner and thus scale linearly to the number of resources which are available and jobs which are run. Network usage is limited to the transfer of input and output files for jobs which execute on cluster nodes. The data transfer is done from the applications to the PlayMolecule backend which in turn stores the files in the MinIO server. Output files are typically in the order of a few megabytes, however applications such as SimpleRun and AdaptiveSampling generate molecular dynamics trajectories of multiple gigabytes and thus can increase network traffic. Computation time varies between applications ranging from a few seconds in ProteinPrepare to multiple weeks in AdaptiveSampling.
Ongoing work in PlayMolecule is focused on reducing data duplication between cluster nodes, the MinIO server and the web server to also reduce network load as well as storage requirements. All PlayMolecule singularity applications are built using internal Python libraries which we regularly evaluate for performance and algorithms.
Clinical Use:
In Silico Trials
License type:
Exposed as paid SaaS; free version with limited capability.
Jimenez-Luna J et al. 2018, KDEEP: Protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. Journal of Chemical Information and Modeling. DOI
Jimenez-Luna J et al. 2019, DeltaDelta neural networks for lead optimization of small molecule potency. DOI
Ferruz N. et al. 2018, Dopamine D3 receptor antagonist reveals a cryptic pocket in aminergic GPCRs. DOI
Lovera S et al. 2019, Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. DOI
Jimenez-Luna J et al. 2019, PathwayMap: Molecular Pathway Association with Self-Normalizing Neural Networks. DOI
Harvey MJ, De Fabritiis G 2009, An Implementation of the Smooth Particle Mesh Ewald Method on GPU Hardware. DOI
Harvey MJ et al. 2009, ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. DOI
For more information about the applications supported in CompBioMed, you can contact us at "software at compbiomed.eu".